Investigadores de las universidades de Bristol, Manchester, Exeter y Newcastle entrenaron el modelo con datos proteómicos de plasma sanguíneo del UK Biobank y lo evaluaron en muestras de saliva de 156 participantes, obteniendo un área bajo la curva de 0.88.
Investigadores de las universidades de Bristol, Manchester, Exeter y Newcastle entrenaron el modelo con datos proteómicos de plasma sanguíneo del UK Biobank. En un estudio publicado en npj Digital Medicine presentaron un marco de aprendizaje profundo o deep learning capaz de detectar cáncer de cabeza y cuello a partir de perfiles proteómicos en saliva, sin haber sido entrenado con ese tipo de muestra. El enfoque combina transferencia entre tipos de muestras biológicas y generación de datos sintéticos para superar desafíos recurrentes en el desarrollo de biomarcadores para cánceres poco frecuentes como el tamaño reducido de los conjuntos de datos y el desequilibrio entre casos y controles.
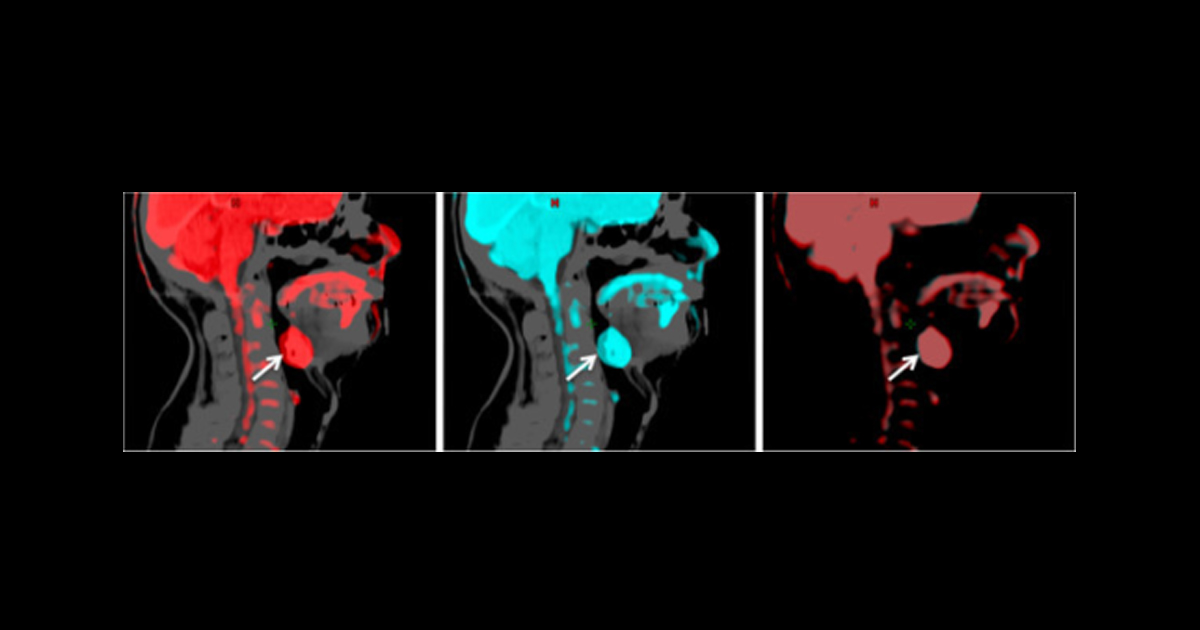
El modelo, denominado CNN-Synth, fue entrenado con datos proteómicos de plasma sanguíneo de 13,208 casos pancancerígenos y 39,806 controles del UK Biobank, una cohorte poblacional que midió 2,941 proteínas en más de 53 mil individuos. Para compensar el desequilibrio entre el número de casos y controles, los investigadores entrenaron un autoencoder variacional (VAE, en inglés) capaz de generar 10 mil perfiles proteicos sintéticos de cáncer, que se sumaron al conjunto de entrenamiento. El modelo resultante fue evaluado en el estudio SensOrPass, un conjunto independiente de 156 participantes, 64 con cáncer de cabeza y cuello y 92 controles, en quienes se midieron 92 proteínas en muestras de saliva.
El estudio mostró que CNN-Synth alcanzó un área bajo la curva (AUC, en inglés) de 0.88, frente a 0.77 del modelo entrenado sin datos sintéticos el CNN-Raw. La reducción en errores de clasificación fue considerable, ya que los falsos positivos disminuyeron de 17 a 8 y los falsos negativos de 25 a 18. Ambos modelos fueron capaces de detectar casos en todos los estadios tumorales, incluyendo enfermedad en etapa temprana, aunque la tasa de error fue más alta en el estadio III para los dos. CNN-Synth mantuvo su ventaja sobre CNN-Raw también al aplicar ajuste fino sobre los datos de saliva mediante validación cruzada estratificada.
Asimismo, al comparar CNN-Synth con una serie de modelos convencionales no neuronales, incluyendo regresión logística, análisis discriminante lineal, redes de vecinos más cercanos, árboles de decisión, máquinas de soporte vectorial y XGBoost, todos entrenados con los mismos datos del UK Biobank más los casos sintéticos, la red convolucional superó a todos en el escenario de transferencia entre tipos de muestra. XGBoost fue el de mejor desempeño entre los métodos no neuronales, con un AUC de 0.69.
Para interpretar qué proteínas contribuyeron más a las predicciones del modelo, los investigadores aplicaron el método SHapley Additive exPlanations (SHAP). En CNN-Synth, las proteínas con mayor peso fueron IL6, CXCL17, CXCL13, IGF1R y FASLG, todas con roles documentados en biología del cáncer. El análisis de enriquecimiento funcional de las 20 proteínas con mayor valor SHAP mostró representación significativa en procesos relacionados con la progresión tumoral, incluyendo morfogénesis, diferenciación de células inmunes, desarrollo vascular, remodelación de la matriz extracelular y vías de señalización.
No obstante, los autores señalan varias limitaciones, principalmente con la cohorte de prueba, la cual es pequeña y étnicamente homogénea, con 98.9% de participantes de origen británico blanco, lo que restringe la generalización del modelo a otras poblaciones. De igual forma, este fue entrenado con todos los tipos de cáncer disponibles en el UK Biobank, dado que los casos específicos de cáncer de cabeza y cuello representan solo el 14% del total, lo que implica que los patrones aprendidos reflejan señales pancancerígenas más que características propias de este tumor. Además, los autores tampoco exploraron si arquitecturas más simples podrían haber logrado resultados similares, ni contaron con mediciones de otras moléculas como expresión génica o metilitos que podrían haber mejorado el desempeño.