Investigadores de NYU Langone se encuentran desarrollando una estrategia más realista para estudiar la enfermedad de Hirschsprung.
Científicos de NYU Langone Health desarrollaron un nuevo enfoque experimental que permite estudiar con mayor precisión la enfermedad de Hirschsprung (HSCR), un trastorno congénito poco frecuente que afecta el intestino de recién nacidos y puede provocar obstrucciones graves al impedir el paso normal de las heces.
Durante el desarrollo del sistema digestivo, se forma una compleja red de nervios conocida como sistema nervioso entérico, a menudo descrita como un “segundo cerebro”, encargada de regular el movimiento de los alimentos y los desechos a través del intestino. En la HSCR, alteraciones genéticas impiden que esta red nerviosa se desarrolle correctamente, lo que da lugar a zonas del intestino sin inervación.
Hasta ahora, la mayoría de los modelos animales para estudiar esta enfermedad se basaban en la eliminación completa de un solo gen relacionado con la HSCR. Aunque estos modelos lograban reproducir algunos aspectos del trastorno, no reflejaban con fidelidad cómo se manifiesta en las personas.
Por ejemplo, en humanos la enfermedad es aproximadamente cuatro veces más frecuente en varones y suele afectar principalmente la parte final del colon. Sin embargo, en los modelos tradicionales en ratones, la incidencia era similar entre machos y hembras, y las alteraciones nerviosas se extendían por todo el intestino. El nuevo estudio, publicado en Proceedings of the National Academy of Sciences (PNAS), propone un enfoque distinto a través del análisis de la interacción entre múltiples mutaciones genéticas, en lugar de estudiar un solo gen de forma aislada.
El trabajo fue liderado por el Dr. Aravinda Chakravarti, investigador con más de tres décadas de experiencia en el estudio de la HSCR, quien ayudó a identificar dos de los principales genes implicados en esta condición RET y EDNRB.
En lugar de eliminar por completo estos genes, los investigadores crearon combinaciones de mutaciones más leves, en las que uno o ambos genes conservaban parte de su función. La combinación que mejor reprodujo la enfermedad humana fue aquella en la que solo una copia del gen RET estaba inactiva y ambas copias de EDNRB seguían siendo parcialmente funcionales.
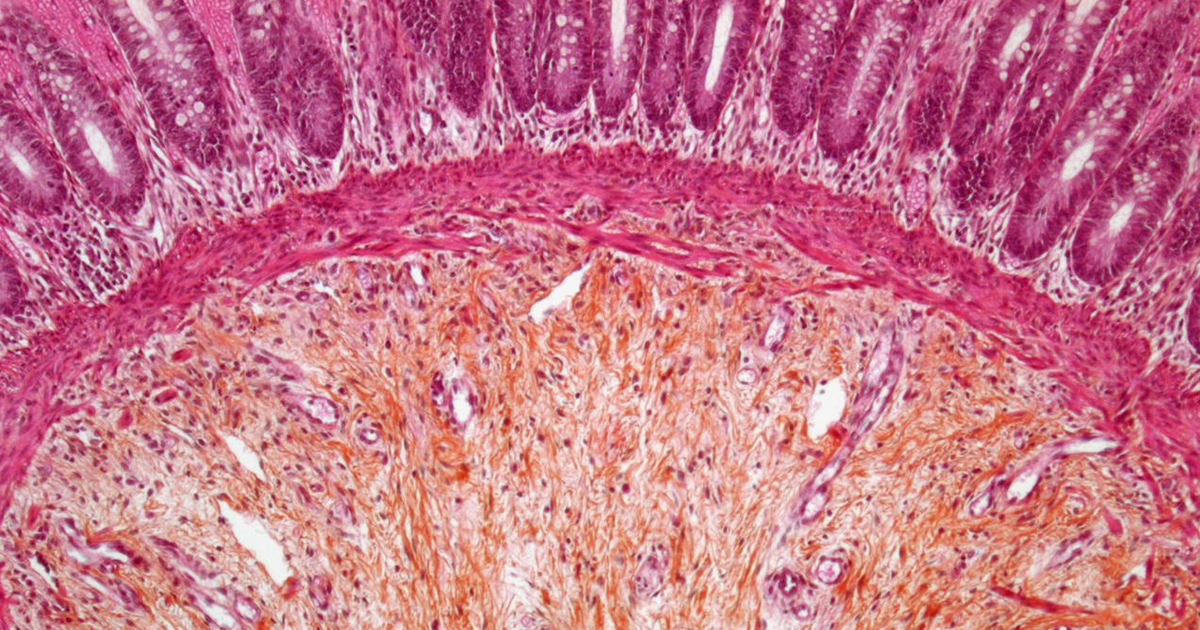
Estos ratones mostraron un desarrollo nervioso normal en el intestino delgado, una afectación limitada al colon y una mayor prevalencia de la enfermedad en machos, características que se asemejan mucho más a las observadas en pacientes humanos.
Al analizar el desarrollo intestinal de estos ratones, los investigadores hicieron un hallazgo inesperado. Aunque la enfermedad se asocia con la ausencia total de nervios en ciertas zonas del intestino, durante el desarrollo embrionario los animales presentaban una abundancia de células nerviosas inmaduras, incluso mayor que en ratones sanos.
El análisis genético reveló un aumento significativo en la actividad de SOX2OT, un gen implicado en la maduración de las células precursoras del sistema nervioso. Los científicos plantean que, sin la regulación adecuada de RET y EDNRB, este gen podría interferir con el proceso de maduración celular, impidiendo que las neuronas intestinales se desarrollen completamente.
Según los autores, este nuevo modelo permitirá responder preguntas clave sobre los mecanismos de la HSCR y explorar posibles estrategias terapéuticas. Además, destacan que el enfoque podría aplicarse a otros trastornos complejos del desarrollo, que no suelen depender de una sola mutación genética, sino de la interacción de múltiples cambios pequeños en distintos genes.
“Estudiar las enfermedades complejas tal como ocurren en los seres humanos —como el resultado de múltiples mutaciones parciales y no de la pérdida total de un solo gen— nos acerca a comprender mejor sus matices y, eventualmente, a desarrollar tratamientos que salven vidas”, señaló el Dr. Chakravarti.





