Este nuevo enfoque combina datos evolutivos y de población humana para identificar mutaciones que podrían causar enfermedades raras.
Investigadores de Harvard Medical School presentaron popEVE, un modelo computacional capaz de estimar qué tan perjudicial puede ser una variante genética en cualquier proteína del cuerpo humano. El avance, publicado en Nature Genetics, propone una forma más precisa y comparable de analizar mutaciones relacionadas con enfermedades, especialmente aquellas que afectan el desarrollo infantil y para las cuales la interpretación genética sigue siendo compleja.
La interpretación de variantes missense, es decir, una mutación genética en la que el cambio de un solo nucleótido en la secuencia de ADN resulta en un codón que especifica un aminoácido diferente. La interpretación de variantes missense, que cambian un solo aminoácido en una proteína, continúa siendo uno de los mayores desafíos de la genética clínica. Su efecto suele depender del contexto y no siempre es evidente si una mutación es dañina o si impacta de forma distinta según el gen afectado. Aunque los modelos actuales ofrecen buenos resultados en genes ya estudiados, no permiten comparar fácilmente la gravedad de variantes ubicadas en distintas regiones del genoma, lo que limita su uso en diagnósticos complejos.
“Nuestro objetivo era desarrollar un modelo que clasificara las variantes según la gravedad de la enfermedad, proporcionando una visión priorizada y clínicamente significativa del genoma de una persona”, detalló la coautora principal Debora Marks, profesora de biología de sistemas en el Instituto Blavatnik de Harvard.

Para enfrentar este problema, el equipo desarrolló popEVE, un modelo que integra información evolutiva, proveniente de secuencias de proteínas en múltiples especies, con datos de variación genética en grandes poblaciones humanas. Al combinar ambas fuentes, el modelo produce un puntaje continuo que refleja la severidad potencial de una variante y, al mismo tiempo, hace que esos valores sean comparables entre diferentes proteínas, una característica esencial para la interpretación clínica que no estaba disponible en métodos previos.
Los resultados del estudio muestran que popEVE es capaz de diferenciar con mayor eficacia variantes asociadas a trastornos graves de aparición temprana frente a aquellas relacionadas con síntomas más moderados o tardíos. En una cohorte de más de 31,000 personas con trastornos del desarrollo, el modelo identificó variantes en 442 genes potencialmente implicados, incluyendo 123 genes candidatos no reportados previamente. Muchas de estas variantes se localizaron en zonas críticas de las proteínas, cercanas a superficies de interacción con otras moléculas, lo que respalda su posible impacto en funciones biológicas esenciales.
“Se trata de enfermedades que, por su gravedad, suponíamos que eran genéticas y causadas por una única variante, pero dicha variante no se había encontrado”, afirmó Rose Orenbuch, de Mark Labs y autora principal del estudio.
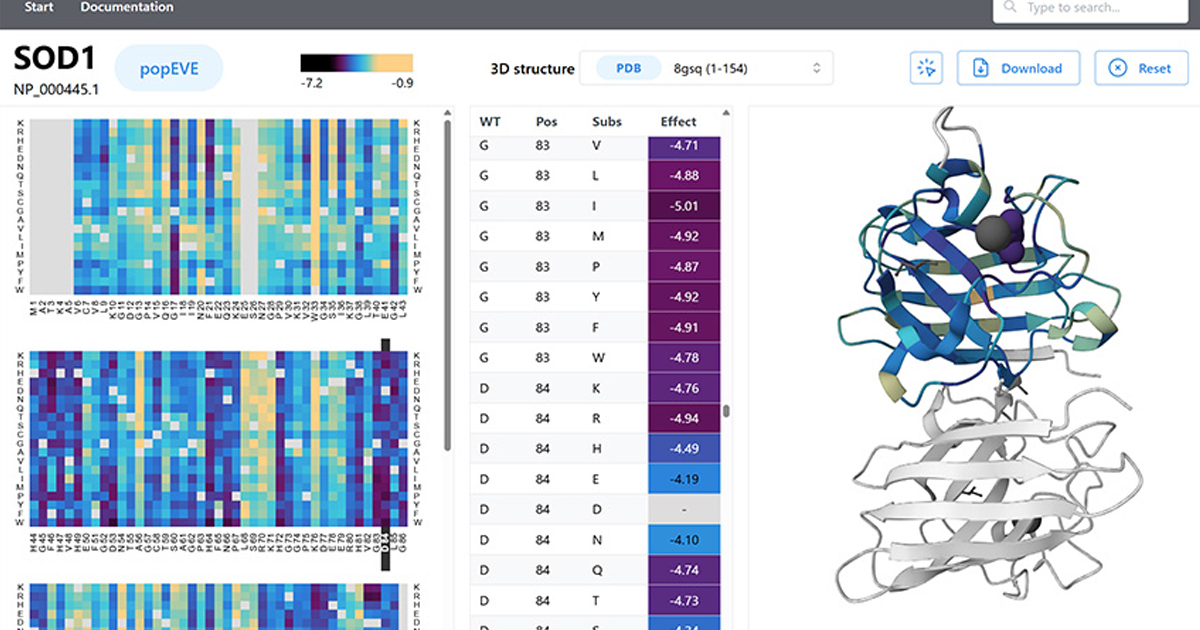
El estudio también destaca que popEVE puede priorizar variantes causales utilizando únicamente el exoma del paciente, sin necesidad de contar con la secuenciación de los padres. Esto representa una ventaja para entornos clínicos en los que no siempre es posible obtener información genética familiar y abre la puerta a diagnósticos más accesibles en casos de enfermedades raras.
De acuerdo con los autores, este enfoque tiene el potencial de mejorar significativamente el análisis de variantes en estudios clínicos y acelerar la identificación de genes relacionados con enfermedades graves. No obstante, reconocen que aún no existen modelos capaces de evaluar de manera conjunta mutaciones missense y variantes de pérdida de función, un siguiente paso necesario para avanzar hacia herramientas más completas. El estudio subraya también la necesidad de desarrollar métodos más eficientes desde el punto de vista computacional, dado el alto costo energético y económico de entrenar modelos de gran escala.